
Ehlers–Danlos sindromi
Bu maqolada bir qancha muammolar mavjud. Iltimos, ularni tuzatib yordam qiling yoki shu muammolarni munozara sahifasida muhokama qiling.
|
Ehlers-Danlos sindromi — biriktiruvchi toʻqimalarning genetik patologiyalari guruhiga kiradi. Bunda kollagen tuzilishini kodlovchi DNK sohalarida yoki kollagen tolasi hosil boʻlish jarayonida qatnashuvchi biologik faol oqsillar haqida maʼlumot saqlovchi DNK qismlaridagi patologiya hisoblanadi.
Kasallikning aniqlangan shakllarining tarqalishi 1:15 000 tugʻilishni tashkil etadi, ammo haqiqiy tarqalish ancha yuqori, bu kasallikning tekshirish qiyinligi va kasallik kechishining koʻplab yengil va yashirin variantlari mavjudligi bilan bogʻliqdir. Ogʻir shakllari juda kam uchraydi, ularning chastotasi 1:100 000 tugʻiladi.
.png)
Boʻgʻimlarning boʻshashishi, terining haddan tashqari choʻzilishi, atrofik chandiqlar paydo boʻlishi bilan yaraning yomon bitishi bu kasallikning asosiy xarakterli belgilari boʻlib sanaladi.
Tarixi
[tahrir | manbasini tahrirlash]Birinchi marta 1682-yilda Job Van Makaren tomonidan tasvirlangan, keyinchalik esa ushbu sindrom Edvard Ehlers va Genri-Aleksandre Danlos tomonidan 1901 va 1908-yillarda nashr etilgan oʻzlarining ishlarida batafsil tahlil qilingan.
Patogenezi
[tahrir | manbasini tahrirlash]Ehlers-Danlos sindromi — bu irsiy va kimyoviy anomaliyada farq qiluvchi biriktiruvchi toʻqima displaziyalarining kombinatsiyasidir.
Tasdiqlangan kuzatuvlarning koʻpchiligida autosomal dominant tarzda nnasldan — naslga oʻtishi qayd etilgan.
Bu sindromda bilan biriktiruvchi toʻqimaning asosiy elementlaridan biri — kollagen tolasining asosini tashkil etuvchi fibillan oqsilining tuzilishini kodlovchi genlarning DNK tuzilishida mutatsiyalar kuzatiladi. Kasallikning birinchi va ikkinchi turlarida fibroblast faolligining pasayishi, proteoglikanlar ishlab chiqarilishining koʻpayishi, kollagenning normal ishlab chiqarilishiga hissa qoʻshadigan oqsillarning faolligining pasayishi yoki toʻliq yoʻqligi kuzatiladi. Proalfa 1 (V) yoki Proalfa 2 (V) kollagen zanjirlari beshinchi tipida molekulyar nuqson topilgan. Autosomal — dominant tipda irsiylanadi. Uchinchi tip kollagen III alfa1, tenascin X sintezi uchun javobgar genlardagi mutatsiyalar tufayli yuzaga keladi, bu kollagen ishlab chiqarishning yetarli emasligi bilan namoyon boʻladi bu esa oʻz navbatida boylamlar va paylarning shikastlanishi va tez-tez boʻgʻimlarning joyidan chiqib ketishiga olib keladi. Bu turi ham autosomal -dominant turda irsiylanadi. Toʻrtinchi tur yurak-qon tomir tizimining patologiyasi bilan namoyon boʻladigan qon tomir devori tuzilishining bir qismi boʻlgan uchinchi turdagi kollagen tolalari ishlab chiqarilmasligi bilan belgilanadi va teri kasallikning boshqa kichik tiplariga qaraganda kamroq shikastlanadi. Barcha kalibrli tomirlarning tez-tez yorilishi kuzatiladi.
Beshinchi variantda X xromosomasi bilan bogʻliq boʻlgan retsessiv irsiylanishi tasvirlangan, uning patogenezi yetarlicha oʻrganilmagan.
Oltinchi turida kollagen tarkibida gidroksil guruhining lizinga biriktirilishini taʼminlovchi, yaʼni kollagenni oʻzgartiruvchi ferment boʻlgan lizilgidroksilaza oqsilining etishmasligi bilan bogʻliq. Autosoma — retsessiv ravishda irsiylanadi. Bu nafaqat pay, bogʻlamlar va dermaning shikastlanishi, balki koʻruv va mushak apparatlarining patologiyasi bilan ham namoyon boʻladi.
Yettinchi tipda birinchi turdagi kollagen prekursorining kollagenga oʻzgarishi buziladi. Irsiylanishining autosomal — retsessiv va autosomal — dominant variantlari tasvirlangan. Bunday bemorlar qisqa boʻyli boʻlib, ular ogʻir qoʻshma patologiyaga ega boʻladilar.
Oʻninchi tipda hujayralararo moddaning hosil boʻlishida ishtirok etadigan plazma fibronektinida nuqson mavjud boʻladi. Kasallikning klassik koʻrinishlaridan tashqari, oʻninchi tipda trombotsitlarning agregatsiya xususiyatlarining buzilishi bilan bogʻliq boʻlgan qon ivishining buzilishi va terida chiziqli chandiqlar koʻzga tashlanadi.[3] Autosomal — retsessiv turda irsiylanadi.
Patologik jihatdan barcha turdagi Ehlers-Danlos sindromi teri qalinligining pasayishi, kollagen tuzilmalarining patologik yoʻnalishi va strukturasining buzilishi, elastik tolalarning ustunligi, gipervaskulyarizatsiya, tomirlar va arteriyalar diametrining oshishi bilan namoyon boʻladi.

Belgilari
[tahrir | manbasini tahrirlash]- terining elastikligini oshishi
- teri ostida qizil tusda halqalarning boʻlishi
- boʻgʻimlarning giperharakatchanligi
- biriktiruvchi toʻqima tuzilmalarining zaifligi;
- qon ketishining kuchayishi.
Kasallikning klinik koʻrinishi sezilarli darajada sindromning turi va kichik turiga bogʻliq.

1-tip quyidagi klinik koʻrinishga ega:
- terining haddan tashqari choʻziluvchanligi;
- keng atrofik chandiqlar;
- boʻgʻim tizimning harakatchanligini oshirish;
- silliq, baxmal teri;
- teri ostidagi tugunchalar;
- teri ostidagi yumaloq oʻsmasimon hosilalar;
- boʻgʻimlarning zaifligi belgilari;
- oyoq kaftining tekislanishi — yassioyoqlik;
- minimal taʼsirga ega gematomalar;

- diafragma churrasi;
- tos boʻshligʻi mushaklarining zaifligi;
- ayollarda bachadon boʻyni yetishmovchiligi;
- operatsiyalardan soʻng yumshoq toʻqimalar nuqsonlarining shakllanishi.

Giperharakatchan (gipermobil) shaklida
- terining haddan tashqari choʻzilishi
- boʻgʻimlarning takroriy dislokatsiyasi(oʻz joyidan siljishi)
- muntazam artralgiya va mialgiyalar bilan namoyon boʻladi.
Qon tomir shaklida
- akrogeriya (qoʻl va oyoq teri qoplamida degenerativ oʻzgarishlar),

- mayda boʻgʻimlarda harakatchanlikning kuchayishi
- tayanch-harakat tizimining yumshoq toʻqimalarining yorilishi,
- maymoqoyoqlik
- yoshlikdanoq vena tomirlari varikoz kengayishi kuzatiladi
- pnevmo- va pnevmogemotoraks
- milklarning toʻliq rivojlanmaganligi

Kifoskoliotik shakl

- neonatal davrdayoq mushak tonusining keskin pasayishi
- vaqt oʻtishi bilan yomonlashadigan tugʻma skolioz
- skleraning yupqalashishi
- koʻzning yorilishi
- terining tuzilishining buzilishi
- koʻkarishlar, tomirlarning yorilishi
- marfanoid koʻrinishi
- diametri yetarli boʻlmagan shox parda
- rentgenografiya paytida aniqlanadigan suyak shakllanishining buzilishi kuzatiladi.
Artrokalaziya shakli quyidagi klinik koʻrinishlarga ega:
- boʻgʻimlarning tez -tez joylaridan siljib ketishi
- neonatal davrdayoq aniqlanadigan har ikki tomondagi son -chanoq boʻgʻimining notoʻgʻri joylashuvi
- terining giperelastikligi;
- toʻqimalarning zaifligi;
- striae(chiziqlar, terida ingichka toʻlqinsimon chiziqlarning) shakllanishi, minimal taʼsir qilish koʻrsatilsa qon ketishi;
- mushaklarning tarangligining pasayishi
- suyak mineral zichligining pasayishi;
- umurtqa pogʻonasining egriligi.
Dermatosparaksik shaklda
- terining sezilarli atrofiyasi, osilishi,
- ortiqcha teri qoplami
- dermaning yumshashi
- oson qon ketishi
- homiladorlik paytida istmik-servikal etishmovchilik
- katta churralar kuzatiladi.
Tasnifi va rivojlanish bosqichlari
[tahrir | manbasini tahrirlash]Eski tasnifga koʻra, EDMSning 11 turi ajralib turadi (hozirda IX va XI tasnifdan chiqarib tashlangan). 1-toifa — ogʻir, klassik; 2-toifa — yengil, klassik; 3-toifa — yaxshi sifatli shakli, barcha boʻgʻimlarning harakatchanligi kuchaygan, mushaklar va skeletning patologiyasi yoʻq, teri deyarli patologik jarayonlarga duch kelmaydi; 4 turi — arterial yoki ekximotik (tomir)shakli; 5-toifa — boʻgʻimlarning minimal giperharakatchanligi, terining sezilarli darajada yuqori choʻzilishi. Dermaning gemorragik koʻrinishlari va nuqsonlari oʻrtacha; 6-toifa — okulo-skoliotik, umurtqa pogʻonasining jiddiy egriligi, osteoporoz, teri va artikulyar apparatlarning patologik jarayonga ozgina aralashishi, koʻz toʻqimalarining sezilarli zaifligi, koʻz olmasining yorilishiga olib kelishi mumkin; 7-toifa — bemorlarning boʻyi pastligi, boʻgʻimlarning sezilarli umumiy gipermobilligi, katta boʻgʻimlarning boylamlarining zaifligi bilan tavsiflanadi. Terining choʻzilishi biroz oshadi. Yuzning xarakterli xususiyatlari: keng joylashgan koʻzlar, epikant, yuz oʻrta qismining pachoqligi; 8-toifa — boʻgʻimlarning gipermobilligi, terining sezilarli zaifligi bilan namoyon boʻladi. Periodontal kasallik tufayli koʻpincha tishlarning erta yoʻqolishi yuzaga keladi; 10-toifa — sindromning tipik koʻrinishlariga qoʻshimcha ravishda trombotsitlarning gipoagregatsiyasi mavjud. Terida atrofik chandiqlar va koʻkarishlar aniqlanadi. Piter Bayton va boshqalar tomonidan soddalashtirilgan klinik tasnifida kasallikning oltita asosiy turi mavjud. Aynan shu tasnif xalqaro foydalanish uchun eng maqbul hisoblanadi. Shakllarning mutlaq soni uchun katta va kichik diagnostika mezonlari mavjud. Kasallikni maʼlum bir turga bogʻlash uchun kamida 1 ta asosiy mezon talab qilinadi. Kichik mezonlar kasallikning pastki turini aniqlashga yordam beradi, ammo tashxis qoʻyish uchun bir oʻzi yetarli emas.
Klassik turi
Katta mezonlar:
- teri qoplamlarining haddan tashqari tarangligi;
- chiziqlar — striyalar;
- bogʻlamlarning haddan tashqari choʻzilishiga olib keladigan harakatchanlikni oshishi;
- suyaklarning dislokatsiyasi yaʼni notoʻgʻri joylashuvi;
- oyoq kafti gumbazining tekislanishi.
Kichik mezonlar:
- yumshoq, mayin teri;
- soxta oʻsmalar (, teriga tez-tez taʼsir qilish joylarida va shikastlanish joyida teri ostida hosilalar oʻsishi);
- teri osti tugunlari (kichik, harakatchan, zich, distal sohalarda joylashgan, baʼzan dekalsifikatsiyalangan va rentgenogrammalarda koʻrinadigan boʻladi);
- skelet mushaklari gipoplaziyasi;
- koʻkarishlar shakllanishiga moyillik;
- diafragmaning churrasi, tos boʻshligʻi mushaklarining zaifligi bilan namoyon boʻladigan biriktiruvchi toʻqima tuzilmalarining zaifligi;
- operatsiyadan keyingi yumshoq toʻqimalarning nuqsonlari;
- mitral qopqoq nuqsonlari;
- aorta ildizining kengayishi;
- amniotik suyuqlikning muddatidan oldin yorilishi.
Autosomal — dominant tipda irsiylanadi, EDSning klassik turi genetik jihatdan geterogendir: bunda 5-toifa kollagenni kodlovchi genlarda (COL5AI va COL5A2) va 1-toifa kollagenni kodlovchi COL1A1 genda nuqsonlar aniqlanadi.
Klassik turda ikkita kichik tip 1 (ogʻir) va II (yengil) farqlanadi. Aslida esa bu bir xil kasallikning allel shakllaridir.
Teri-boʻgʻim turi
Katta mezonlar:
- choʻziluvchan yoki yumshoq teri;
- barcha boʻgʻimlarning boʻshligi.
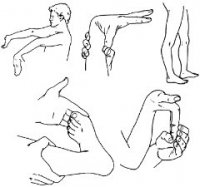

Kichik mezonlar:
- takroriy dislokatsiyalar;
- surunkali artralgiya;
- oʻxshash alomatlarga ega qarindoshlarning mavjudligi;
- mitral qopqoq yetishmovchiligi;
- aorta ildizining kengayishi.
Autosomal — dominant tipda nasldan- naslga oʻtadi.
Qon tomir turi
Katta mezonlar:
- nozik teri;
- qon tomirlarining yorilishi;
- tos boʻshligʻi mushaklarining zaifligi;
- koʻkarishlarga moyillik;
- tipik yuz (nozik uchli burun, kichkina lablar, teri choʻzilgan, yonoqlari choʻkib ketgan va teri osti toʻqimalarining atrofiyasi tufayli koʻzlar boʻrtib ketganga oʻxshaydi).
Ikki yoki undan ortiq asosiy mezonlarning mavjudligi kasallikning ushbu turini tasdiqlashni koʻrsatadi.
Kichik mezonlar:
- kichik boʻgʻinlarning boʻshligi;
- boylam va mushak apparatlarining shikastlanishi;
- siydik pufagining yorilishi;
- vena tomirlarining varikoz kengayishi;
- pnevmo- yoki pnevmogemotoraks;
- tishlarning boʻyinlari va ildizlarining koʻchishi;
- oilada shu kasallik qayd etilganligi;
Autosomal — dominant tipda nasldan- naslga oʻtadi. Kasallik III tipdagi kollagen pro-alfa 1 zanjiri biosintezi haqidagi maʼlumotlarni kodlaydigan COL3A1 genidagi nuqsonlar tufayli yuzaga keladi.
Artikulyar turi (artroxolaziya)
Katta mezonlar:
- koʻpchilik boʻgʻimlarning kuchli boʻshligi;
- takroriy boʻgʻim chiqishlari;
- sonning tugʻma dislokatsiyasi.
Kichik mezonlar:
- terining haddan tashqari choʻzilishi, toʻqimalarning zaifligi;
- koʻkarishlarga moyillik;
- mushaklarning gipotenziyasi;
- kifoskolioz;
- osteoporoz.
Autosomal — dominant tipda nasldan- naslga oʻtadi. Kasallik 1-toifa kollagenning alfa 1 va alfa 2 zanjirlarini kodlovchi COL1A1 va COL1A2 genlarining patologiyasi tufayli yuzaga keladi.
Teri turi (dermatosparaksiya)
Katta mezonlar:
- terining tez shikastlanuvchanligi;
- burmali teri.
Kichik mezonlar:
- yumshoq teri;
- koʻkarishlarga moyillik;
- amniotik suyuqlikning prenatal yorilishi;
- kindik va chov churrasi.
Autosomal — retsessiv tarzda irsiylanadi. Bemorlar geterozigota yoki gomozigota boʻlishiga qaramay ularning barchasida prokollagen-N-endopeptidazani kodlovchi ADAMTS2 genidagi mutatsiya boʻladi. Terining tez shikastlanishi va koʻkarish tendentsiyasiga qaramay, yaraning bitishi normal tarzda sodir boʻladi.
Kifoskoliotik turi
Katta mezonlar:
- koʻpchilik boʻgʻimlarning boʻshligi;
- yangi tugʻilgan chaqaloqlarda kuchli mushak gipotenziyasi;
- jadal rivojlanuvchi tugʻma skolioz;
- skleraning zaifligi va koʻz olmasining yorilishi.
Tashxisni tasdiqlash uchun chaqaloqlarda 3 ta asosiy mezon qayd etilishi kerak.
Kichik mezonlar:
- toʻqimalarning shikatlanishga moyilligi;
- koʻkarishlarga moyillik;
- arteriya yorilishi;
- marfanga oʻxshash koʻrinish;
- mikrokornea — shox pardaning diametrining sezilarli ravishda kamayishi bu esa oʻz navbatida glaukomaga sabab boʻladi;
- osteoporoz.
Autosomal — retsessiv tarzda irsiylanadi. Kasallik lizil gidroksilaza (PLOD) genidagi mutatsiyaga asoslanadi, bu ferment prokollagenning translatsiyadan keyingi modifikatsiyasini amalga oshiradi.
Koʻpgina bemorlar 20-30 yoshga kelib mustaqil harakat qilish qobiliyatini yoʻqotadilar.
Asoratlari
[tahrir | manbasini tahrirlash]- Ehlers-Danlos sindromining eng koʻp uchraydigan asoratlari yirik tomirlarning yorilishidan katta qon ketishdir.
- Kifoskoliotik tip bilan, ancha erta yoshda, umurtqa pogʻonasi patologiyasi bilan bogʻliq mustaqil harakat funktsiyasi yoʻqoladi.
- Ushbu tur bilan koʻrishning yoʻqolishi koʻpincha koʻzning membranalarini yupqalashi tufayli yuzaga keladi, bu uning yorilishiga olib keladi.
Diagnostikasi
[tahrir | manbasini tahrirlash]EDS diagnostikasi ushbu kasallikning turli shakllari uchun asosiy va kichik mezonlarni aniqlashga asoslanadi.
Tekshiruvlar hajmi kasallikning asosiy klinik belgilari mavjudligi bilan belgilanadi. Oilada kasallik haqida maʼlumot toʻplash va DNK molekulyar tadqiqotlarini oʻtkazish juda muhim.
Toʻgʻri tekshirish uchun maʼlum qoidalarga rioya qilish kerak:
- tashxis kamida 1 ta katta mezon mavjudligini talab qiladi;
- agar tahlilni laboratoriya tasdiqlash mumkin boʻlsa, ikki yoki undan ortiq asosiy mezonlarning mavjudligi kasallikning tasdiqlanishini kafolatlaydi;
- kichik mezon kamroq diagnostik oʻziga xoslik bilan tavsiflangan alomatdir. Kichik mezonlarning mavjudligi sindrom turini aniqlashga yordam beradi;
- agar katta mezonlar aniqlanmasa, kichiklari tashxisni tekshirish uchun yetarli emas. Ehlers-Danlos sindromi boʻlgan bemorlarda kichik mezonlar kattalarga qaraganda tez-tez topilganligi sababli, faqat kichik mezonlarning mavjudligi Ehlersga oʻxshash fenotipni tekshirish imkoniyatini aniqlaydi.
Differentsial diagnostika biriktiruvchi toʻqima displaziyasining boshqa turlari bilan amalga oshiriladi.
Dermatoparaksaziya tashxisini tasdiqlash uchun 1-toifa kollagen elektroforezi proteaz ingibitorlari ishtirokida teri biopsiyasi yoki fibroblast kulturasidan bioptat olinadi
Davosi
[tahrir | manbasini tahrirlash]- EDS bilan ogʻrigan bemorlarni davolash muammosini hal qilishda dori-darmon boʻlmagan davolash masalalarini — rejim, parhez, fizioterapiya, psixoterapiyani rivojlantirishga katta eʼtibor beriladi.
- Bemorlarni dispanser roʻyxatidan oʻtkazish boʻyicha mavjud dasturlarning asosini asosan nomedikamentoz davo tashkil etadi.
- Yetarli jismoniy faoliyat, massaj, fizioterapiya, ortopedik reabilitatsiya va kasbni toʻgʻri tanlash tavsiya etiladi. Gidroterapiya, suzishning yuqori samaradorligi kuzatiladi.
Tegishli koʻrsatkichlar mavjud boʻlganda fizioterapiya qoʻllanadi.
- Magnitoterapiya, induktoterapiya va terapevtik lazer keng qoʻllanadi.
- Skolioz, kifoskolioz bilan ogʻrigan bolalarni fizioterapevtik davolashda ogʻriq zonalari uchun CHYuCH terapiyasi (har kuni 1 marta yoki 2 kunda 1 marta)
- mikroelementlarni kiritish, elektroforetik usullar
- gipotonik mushaklarning diadinamik stimulyatsiyasi yordamida antispazmolitiklar;
- mahalliy ultratovushli davolash, vakuum va qoʻlda massaj kursi, mushaklarni kuchaytirishga qaratilgan terapevtik mashqlar va suzish.
Quyidagilardan iborat sanatoriy-kurort davolash koʻrsatiladi:
- Yengil mashq rejimi. Umurtqa pogʻonasiga ortiqcha yuklarni (ogʻirliklarni koʻtarish, sakrash) qoʻymaslik kerak;
- Orqa mushaklarni boʻshashtiruvchi va tonik massaji
- Balneoterapiya (10-12 daqiqa davomida karbonat angidridli vannalar,
- Pelloidoterapiya — umurtqa pogʻonasi boʻylab
- Jismoniy mashqlar terapiyasi, umurtqa pogʻonasini ortiqcha yuklamaslik uchun orqa va yelka-kamar mushaklarini kuchaytirish uchun maxsus mashqlar toʻplami, ularning baʼzilari yotgan holatda amalga oshiriladi.
- Proteinli dieta buyuriladi. Fizioterapiya kurslari, mashqlar terapiyasi.
Organlar va tizimlarning shikastlanish darajasiga qarab klinik koʻrinishlarni davolash. Dori-darmonlar bilan davolash
- aminokislotalar (karnitin, nutraminolar)
- vitamin (D, C, E, B1, B2, B6 vitaminlari)
- mineral komplekslar (magne B6, kaltsiy-D3 nikomed, magnerot), *xondroitin ogʻiz orqali va mahalliy
- ossein-gidroksiapatit komplekslari (osteokea, osteogenon, kalsimaks), *trofik preparatlar (ATP, riboksin, lesitin, koenzim Q10).
Bu dori vositalari har 12 oyda 2-3 marta, bir oydan bir yarim oygacha davom etadigan kombinatsiyalangan kurslarda qabul qilinadi.
- Tibbiy simptomatik davolash artralgiya, mialgiyani bartaraf etish, qon aylanishini yaxshilash, beta-blokatorlar, adaptogen, sedativ, vegetotrop dorilarni qabul qilishni oʻz ichiga oladi.
Shuni esda tutish kerakki, EDS koʻplab organ tizimlarining kasalligi boʻlib, ushbu patologiyani tashxislash va davolash uchun turli xil tizimli usullarni ishlab chiqish va amaliy qoʻllash talab etiladi.
Oqibati
[tahrir | manbasini tahrirlash]- Oqibati koʻp hollarda yaxshi, birinchi (artropatiyalar tufayli) va oltinchi (qon ketish va qon tomirlarining yorilishi tufayli) turlarida jiddiyroq. *Bolalar jismoniy faollik, tik ish bilan bogʻliq boʻlmagan kasb tanlashga yoʻnaltirilgan boʻlishi kerak.
Profilaktika
[tahrir | manbasini tahrirlash]Oldini olish molekulyar genetik tahlil usuli bilan homilada kasallikni prenatal aniqlashdan, tibbiy sabablarga koʻra homiladorlikni toʻxtatishdan iborat. Kasallikning postnatal profilaktikasi ishlab chiqilmagan.
Asoratlarning oldini olish kasallikni toʻgʻri va oʻz vaqtida tashxislash va uni davolashni oʻz ichiga oladi.
Havolalar
[tahrir | manbasini tahrirlash]- balneoterapija_komu_pokazano_a_komu_net
- gryazelechenie-osnovnye-printsipy-i-effektivnost-metoda
- magnitoterapiya (Wayback Machine saytida 2022-05-18 sanasida arxivlangan)
Manbalar
[tahrir | manbasini tahrirlash]- Kennet L.Djons Nasledstvennie sindromi po Devidu Smitu Izd."Praktika", M., 2011. 1002 s.
- Kozlova C.Nasledstvennie sindromi i mediko-geneticheskoe konsultirovanie. M.:Praktika, 2007, 448 s.
- Barabas AP:Ehlers-Danlos syndrome. Associated with prematirity and premature of foetal membranes:possible increase in incidence. BMJ 2:682, 1966
- Badalyan L.O, Skvorsov I. A. Klinicheskaya elektroneyromiografiya. Moskva. „Meditsina“. — 1986. — 367s
- Sindrom Ehlersa-Danlosa. V kn.: Nasledstvennaya patologiya cheloveka. Pod obщ. Red. Veltiщeva Yu. Ye., Bochkova N. P. Moskva. — 1992. — t.1. — s.100-109
- Barohn R. G. Distal myopathies and dystrophies // Smin. — Neurol. — 1993. — Sep. — 13 (3). — p. 247-55
- Barashnev Yu. I., Shexonin B. V., Semyachkina A. N., Makkaev X. M. Diagnostika sindroma Ehlersa-Danlo u detey. // Voprosi oxrani materinstva i detstva. — 1988. — t.33. — № 11. — s. 59-64
- Delyagin V. M., Naricheva I. A., Pilx A. D. Sindrom Ehlersa — Danlosa u detey // Pediatriya. — 1988. — № 12. — s. 8-15
- Ilina N. A., Naricheva I. A., Averyanov Yu. N., Logunova L. V. Klinicheskaya i geneticheskaya geterogennost sindroma Ehlersa-Danlosa//Jurn. nevropatologii i psixiatrii im. S. S. Korsakova. 1983. № 10. — s.1445- 1449